Dielectric-Driven Modulation of Electronic Structure, Optical Absorption, and Vibrational Force Constants in 2A3M5NP: A DFT and TD-DFT Investigation
DOI:
https://doi.org/10.71229/r0sjc822Keywords:
Theoretical Physics, Density Functional Theory (DFT), Solvent EffectsAbstract
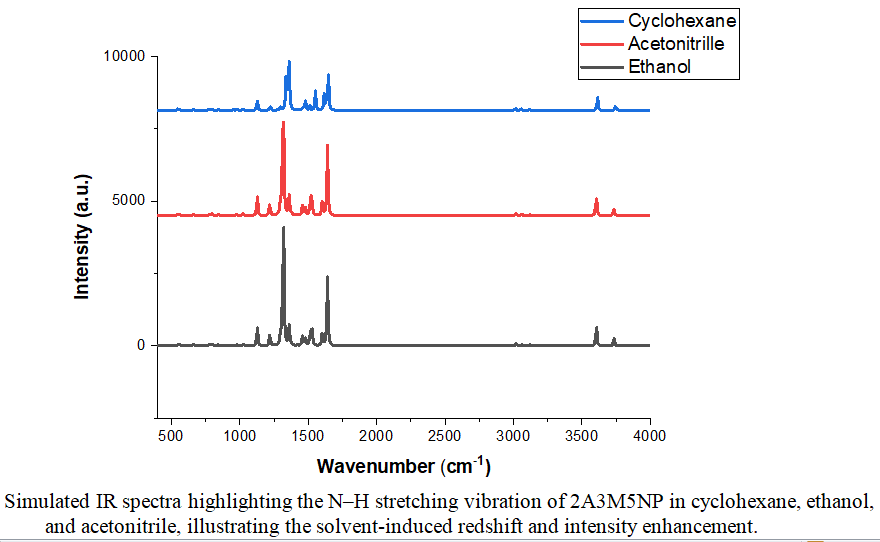
The given work offers a computational study of the effects of dielectric environment on the electronic structure, optical response, and the vibrational behavior of the push pull chromophore 2-amino-3-methyl-5-nitropyridine (2A3M5NP). As part of the IEFPCM method, DFT and TD-DFT calculations at the B3LYP/cc-pVTZ level were used to investigate solvent-dependent variations on the molecular potential energy surface. The frontier orbital distribution is a clear indication of intramolecular charge transfer (ICT) from the amino donor group to the nitro acceptor group with an increase in the solvent polarity increases the Homo-Lumo gap progressively narrows by nearly 6%. This decrease is coupled with a shift in the absorption peak of circa 5 percent in the red direction indicating reflecting stronger stabilization of the excited electronic state in the polar media. It is worth noting that the strong linear correlation that exists between the energy gap and the lambda max also confirms the electronic reorganization of the solvent. The response of the vibrational response also follows the same trend. The N–H stretching mode shows a slight yet measurable of a decrease in the force constant by approximately one-half a percent, leading to a redshift consistent with Hooke's model. Simultaneously, infrared intensity increases due to enhanced dipole moment derivatives on the normal coordinate. When collected, this provides an indication that the electronic and mechanical properties of 2A3M5NP are dielectrically sensitive, which is a physically coherent description of the solvent-selective molecular tuning.
References
] 1] M. Weis, Organic Semiconducting Polymers in Photonic Devices, Applied Sciences, 15 (2025) 4028.https://www.mdpi.com/2076-3417/15/7/4028
[2] S. Sivaprakash, S. Prakash, S. Mohan, S.P. Jose,Quantum chemical studies and spectroscopic investigations on 2-amino-3-methyl-5-nitropyridine by density functional theory,Heliyon, 5 (2019) e02149.https://doi.org/10.1016/j.heliyon.2019.e02149
[3] M. Essid, A. Mezghani, T. Bessaïs, S. Boughzala, Photophysical properties and intramolecular charge transfer in push–pull heterocyclic systems: a theoretical perspective, RSC Advances, 15 (2025) 12345–12360.https://doi.org/10.1039/D5RA06623A
[4] Bui, T.; Nguyen, H. T.; Pham, T. H.; et al. Computational investigation of nonlinear optical properties and intramolecular charge transfer in donor–acceptor systems.Molecules 2023, 28, 1489.https://doi.org/10.3390/molecules28031489
[5] Wang, J.; Gadenne, V.; Patrone, L.; Raimundo, J.-M.Self-Assembled Monolayers of Push–Pull Chromophores as Active Layers and Their Applications.Molecules 2024, 29 (3), 559.https://doi.org/10.3390/molecules29030559
[6] J. M. Herbert, Continuum solvation models: Theory and applications, WIREs Computational Molecular Science, 11 (2021) e1519.https://doi.org/10.1002/wcms.1519
[7] M. Nottoli, G. Lipparini, B. Mennucci,Quantum mechanical treatment of solvent polarization in electronic structure calculations, WIREs Computational Molecular Science, 14 (2024) e1726.https://doi.org/10.1002/wcms.1726 [8] Pliego, J. R., Jr.Reaction Field Contributions to Electronic Energies in Polarizable Continuum Models.The Journal of Physical Chemistry A 2024, 128, 6123–6132.https://doi.org/10.1021/acs.jpca.4c03593
[9] Wang, Y.; Nottoli, M.; Lipparini, F.; Mennucci, B.Electronic Structure Calculations in Polarizable Environments: Advances and Perspectives.Journal of Chemical Theory and Computation 2024, 20 (12), 5210–5231.https://doi.org/10.1021/acs.jctc.4c00594
[10] S. K. Gupta, R. K. Pathak, Solvent polarization effects on electronic structure and optical properties of organic chromophores: A DFT and TD-DFT study, Journal of Molecular Liquids, 382 (2023) 121940.https://doi.org/10.1016/j.molliq.2023.121940
[11] M. U. Khan, S. Nadeem, A. Fatima, J. Yaqoob, M. Khalid, F. Abbas, N. Alhokbany, M. R. S. A. Janjua,DFT molecular simulations for static, dynamic and solvent-dependent nonlinear optical properties of triphenylamine–carbazole-based organic dyes with D–D–A framework, Journal of Molecular Liquids, 391 (2023) 123258.https://doi.org/10.1016/j.molliq.2023.123258
[12] Khan, M. U.; Nadeem, S.; Fatima, A.; Yaqoob, J.; Khalid, M.; Abbas, F.; Alhokbany, N.; Janjua, M. R. S. A.Nonlinear Optical Response and Charge-Transfer Analysis of Donor–Acceptor Chromophores via DFT.ACS Omega 2022, 7 (19), 16500–16512.https://doi.org/10.1021/acsomega.2c01474
[13] Arif, N.; Khan, M. U.; Nadeem, S.; Fatima, A.; Yaqoob, J.; Khalid, M.; Abbas, F.; Al-Sehemi, A. G.; Janjua, M. R. S. A.Theoretical Investigation of Nonlinear Optical Properties in Push–Pull Systems Using DFT Approaches.RSC Advances 2023, 13, 1485–1499.https://doi.org/10.1039/D2RA07134G
[14] Pant, D.; et al.Density Functional Theory Study of Nonlinear Optical Properties and Electronic Structure in Donor–Acceptor Systems.International Journal of Quantum Chemistry 2024, 124, e27317.https://doi.org/10.1002/qua.27317
[15] P. Godlewska et al .Charge-transfer characteristics and nonlinear optical response of organic chromophores: A computational study, International Journal of Molecular Sciences, 26 (2025) 11522.https://doi.org/10.3390/ijms262311522
[16] M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al.,Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009.
[17] A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, The Journal of Chemical Physics, 98 (1993) 5648–5652.https://doi.org/10.1063/1.464913
[18] C. Lee, W. Yang, R. G. Parr,Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density,Physical Review B, 37 (1988) 785–789.https://doi.org/10.1103/PhysRevB.37.785
[19] T. H. Dunning Jr., Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen ,The Journal of Chemical Physics, 90 (1989) 1007–1023.https://doi.org/10.1063/1.456153
[20] P. J. Stephens, F. J. Devlin, C. F. Chabalowski, M. J. Frisch,Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields,The Journal of Physical Chemistry, 98 (1994) 11623–11627.https://doi.org/10.1021/j100096a001
[21] J. Tomasi, B. Mennucci, R. Cammi,Quantum mechanical continuum solvation models,Chemical Reviews, 105 (2005) 2999–3094.https://doi.org/10.1021/cr9904009
[22] M. Cossi, N. Rega, G. Scalmani, V. Barone,Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model,Journal of Computational Chemistry, 24 (2003) 669–681.https://doi.org/10.1002/jcc.10189
[23]M. E. Casida, C. Jamorski, K. C. Casida, D. R. Salahub,Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory,The Journal of Chemical Physics, 113 (2000) 7062–7071.
https://doi.org/10.1063/1.1319643
[24] F. Furche, On the density matrix based approach to time-dependent density functional response theory,The Journal of Chemical Physics, 114 (2001) 5982–5992.https://doi.org/10.1063/1.1353585
[25] A. Dreuw, M. Head-Gordon, Single-reference ab initio methods for the calculation of excited states of large molecules,Chemical Reviews, 105 (2005) 4009–4037.https://doi.org/10.1021/cr0505627
[26] R. Dennington, T. Keith, J. Millam,GaussView, Version 6, Semichem Inc., Shawnee Mission, KS (2016).
[27] OriginLab Corporation,OriginPro 2019b User Guide, Northampton, MA, USA (2019).
[28] A. P. Scott, L. Radom, Harmonic vibrational frequencies: An evaluation of Hartree–Fock, Møller–Plesset, and density functional theory procedures,
The Journal of Physical Chemistry, 100 (1996) 16502–16513.https://doi.org/10.1021/jp960976r
[29] J. P. Merrick, D. Moran, L. Radom,An evaluation of harmonic vibrational frequency scale factors,The Journal of Physical Chemistry A, 111 (2007) 11683–11700.https://doi.org/10.1021/jp073974n
[30] S. Fekadu Mamo, L. Gorfe, T. Gidey, et al., Exploring solvent polarity effects on molecular structure and photophysical properties: A DFT and TD-DFT study,
AIP Advances, 15 (2025) 075328.https://doi.org/10.1063/5.0280338
[31] X. Bai, Y. Liang, Z. Shi, et al.,Solvent-mediated control of intramolecular charge transfer in organic chromophores,Molecules, 31 (2025) 76.https://doi.org/10.3390/molecules31010076
[32] Abdelaziz, B.; et al. DFT-QTAIM Investigation on Twisted Intramolecular Charge Transfer Mechanisms.The Journal of Physical Chemistry A 2023, 127 (39), 8256–8268.https://doi.org/10.1021/acs.jpca.3c04277
[33] Mester, D.; Kállay, M.Charge-Transfer Excitations within Density Functional Theory.Journal of Chemical Theory and Computation 2022, 18 (3), 1486–1497.https://doi.org/10.1021/acs.jctc.1c01307
[34] G. Scalmani, M. J. Frisch,Continuous surface charge polarizable continuum models of solvation. I. General formalism, The Journal of Chemical Physics, 132 (2010) 114110.https://doi.org/10.1063/1.3359469
[35] Bowling, P. E.; Gray, M.; Paul, S. K.; Herbert, J. M.Testing a Heterogeneous Polarizable Continuum Model against Exact Poisson Boundary Conditions.Journal of Chemical Theory and Computation 2025, 21 (4), 1722–1738.https://doi.org/10.1021/acs.jctc.4c01665
[36] Goli, D. R.; Rehman, S.; Khalid, M.; et al.Solvent polarity effects on the FT-IR spectrum and electronic properties including HOMO–LUMO gap in metronidazole.RSC Advances 2025, 15, 28538–28554.https://doi.org/10.1039/D5RA02359A
[37] Rouillon, J., Benitez-Martin, C., Grøtli, M. & Andréasson, J., “Click and shift: the effect of triazole on solvatochromic dyes”, Physical Chemistry Chemical Physics, 27 (2025) 4679–4685. https://doi.org/10.1039/D4CP04642K
[38] Mataga, N.; Kaifu, Y.; Koizumi, M.Solvent Effects upon Fluorescence Spectra and the Dipole Moments of Excited Molecules.Bulletin of the Chemical Society of Japan 1956, 29, 465–470.https://doi.org/10.1246/bcsj.29.465
[39] M. Shafiq, M. Khalid, et al., Unraveling the NLO potential of functionalized organic chromophores: Solvent influence on frontier orbitals and absorption behaviour, Scientific Reports, 15 (2025) 4911.https://doi.org/10.1038/s41598-025-04911-7
[40] Elizabeth Mathew, I. Hubert Joe, Solvent dependent nonlinear optical properties of benzodioxol chalcone: An elucidation of spectroscopic, DFT, solvatochromism and z-scan technique ,Journal of Molecular Liquids, 392 (2023) 123415.https://doi.org/10.1016/j.molliq.2023.123415
[41] Hrivnák, T.; Medveď, J.; Papadopoulos, M. G.; et al. Hyperpolarizabilities of Push–Pull Chromophores in Solution: Interplay between Electronic and Vibrational Contributions. Molecules 2022, 27(24), 8738.DOI: https://doi.org/10.3390/molecules27248738
[42] P. K. Dhanya, Arjun J., Navjot Kaur, R. R. Pillai,Solvent-dependent electronic, photophysical and nonlinear optical properties of azulene-based push–pull chromophores: A DFT approach,Journal of Molecular Graphics and Modelling, 142 (2025) 109180.https://doi.org/10.1016/j.jmgm.2025.109180
[43] S. D. E. Fried, S. Bagchi, S. Gnanakaran,Environment- and conformation-induced frequency shifts of vibrational modes in solution,Journal of Physical Chemistry A, 128 (2024) 400–412.https://pmc.ncbi.nlm.nih.gov/articles/PMC11927941
[44] G. Vengatesh; P. Vilambath; Kalyani M. S.; S. Paroth; A. Sivasankaran,
A comprehensive study on solvatochromism, pH effects, and picric acid sensing properties of novel donor–acceptor pyrrole-3-one derivatives: Experimental and DFT studies, Journal of Molecular Liquids, 418 (2025) 126692.
Downloads
Published
Issue
Section
License
Copyright (c) 2026 Al-Noor Journal of Engineering Management and Computer Science

This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.
How to Cite









